Računalniška kemija: metode, simulacije in načrtovanje zdravil
Računalniška kemija: napredne metode in simulacije za natančno napovedovanje lastnosti molekul ter hitrejše in učinkovitejše načrtovanje zdravil in materialov.
Računalniška kemija je veja kemije, ki pri reševanju kemijskih problemov uporablja računalništvo. Ti programi izračunavajo strukture in lastnosti molekul in trdnih snovi. Računalniška kemija običajno dopolnjuje informacije, pridobljene s kemijskimi poskusi. Z njo je mogoče napovedati kemijske pojave, ki še niso bili opaženi. Pogosto se uporablja pri načrtovanju novih zdravil in materialov.
Računalniška kemija lahko predvidi strukturo (tj. predvidene položaje atomov molekule), absolutne in relativne (interakcijske) energije, porazdelitev elektronskega naboja, dipole in višje multipolne momente, vibracijske frekvence, reaktivnost ali druge spektroskopske količine ter preseke za trke z drugimi delci.
Računalniška kemija obravnava statične in dinamične sisteme. V vseh primerih se z večanjem velikosti preučevanega sistema povečuje tudi porabljeni računalniški čas in drugi viri (kot sta pomnilnik in prostor na disku). Ta sistem je lahko posamezna molekula, skupina molekul ali trdna snov. Metode računalniške kemije so od zelo natančnih do zelo približnih. Zelo natančne metode so običajno izvedljive le za majhne sisteme.
Galerija slik
8 Slike


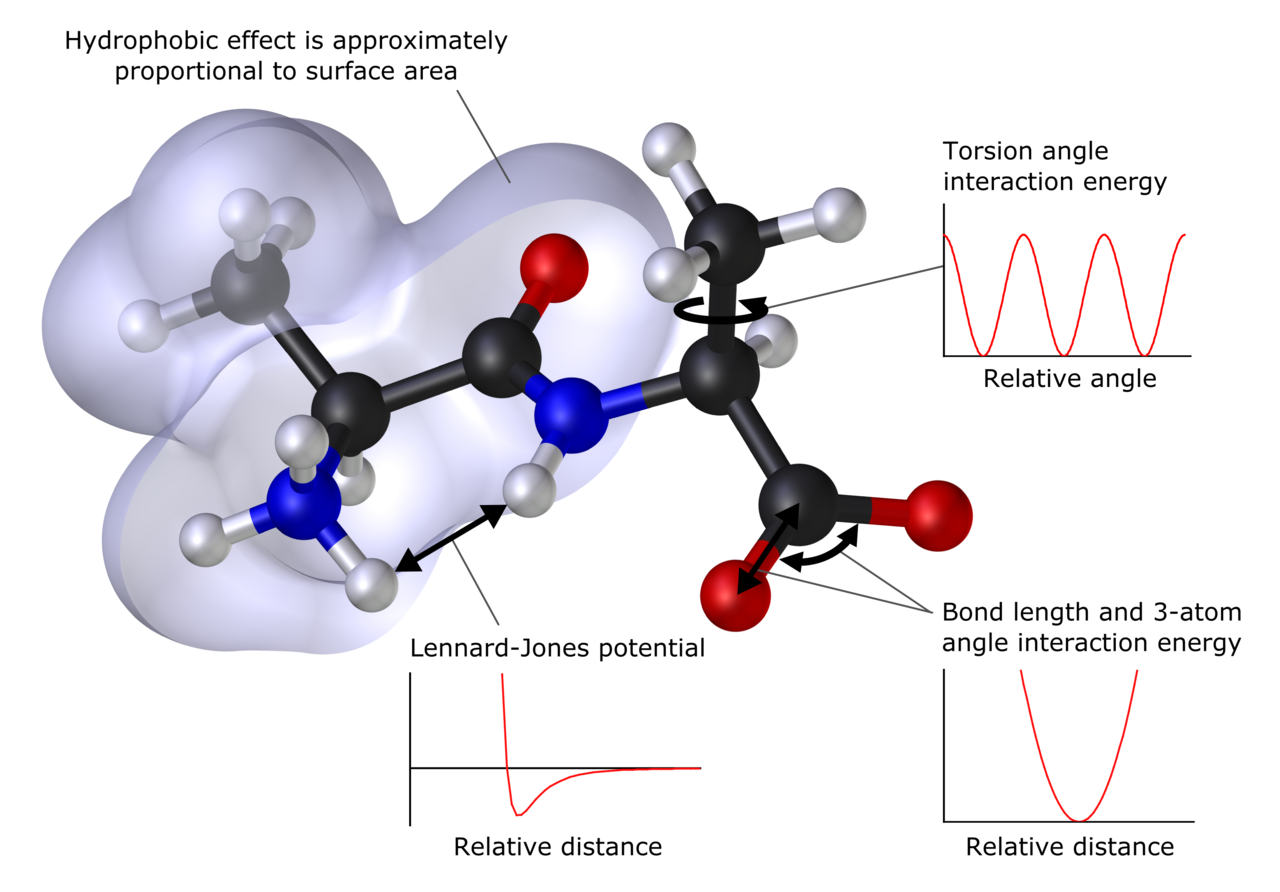


Metode računalniške kemije
Metode se običajno delijo v dve glavni skupini: kvantno-mehanske (QM) in klasične (MM oziroma sile polja). Vsaka ima prednosti in slabosti glede natančnosti, obsega in porabe virov.
- Kvantno-mehanske metode
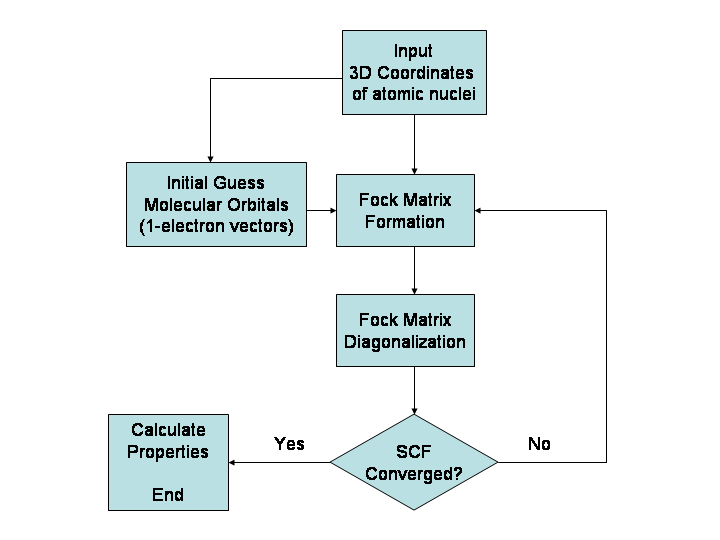
- Ab initio metode (npr. Hartree–Fock, MP2, CCSD(T)) rešujejo Schrödingerjevo enačbo z različnimi približki. So zelo natančne za majhne sisteme, a zahtevajo veliko računalniških virov.
- Teorija funkcionala gostote (DFT) je kompromis med natančnostjo in hitrostjo. Izbira funkcionala (npr. B3LYP, PBE, M06) in baza je ključna za rezultate; v praksi se pogosto dodajajo korekcije za disperzijske interakcije.
- Semi-empirične metode uporabijo poenostavitve in parametre iz eksperimentov, da zmanjšajo zahtevnost izračunov, kar omogoča obdelavo večjih sistemov z manjšo natančnostjo.
- Molekulska mehanika in sile polja
- Molekulska mehanika (MM) uporablja potencialne funkcije (sile polja) za opis vezi, kot so AMBER, CHARMM, OPLS in drude polarizabilni modeli. Primerna je za velike biomolekule in dolge časovne skale.
- Hibridne QM/MM metode kombinirajo natančnost QM (reakcijsko jedro) z učinkovitostjo MM za okolico — pogosto uporabljene pri simulacijah enzymatskih reakcij.
Simulacije in dinamika
Simulacije omogočajo preučevanje gibanja atomov in molekul skozi čas ter ocenitev termodinamičnih in kinetičnih lastnosti.
- Molekulska dinamika (MD) sledi gibanju atomov z reševanjem Newtonovih enačb gibanja. Uporablja se za raziskovanje konformacij, transportnih procesov, interakcij ligand–receptor in lastnosti tekočin ter materialov.
- Monte Carlo (MC) metode vzorčijo prostorsko konfiguracijo z naključnimi premiki in so uporabne za izračun termodinamičnih lastnosti in prostorskih porazdelitev pri ravnotežju.
- Napredne tehnike, kot so pospešeno vzorčenje (umbrella sampling, metadynamics) in izračuni prostih energij (FEP, TI), omogočajo natančno napovedovanje afinitet in reakcijskih poti.
Načrtovanje zdravil in virtualno preskušanje
Računalniška kemija igra ključno vlogo v sodobnem načrtovanju zdravil:
- Virtualno zasukanje in preskušanje (virtual screening) omogoča hitro ocenjevanje velikih knjižnic spojin za vezavo na tarčo.
- Molekulsko dokiranje (docking) predlaga možne položaje vezave liganda v vezavno mesto receptorja in jih ocenjuje s funkcijo točkovanja.
- Prosti-energijski izračuni (FEP/TI) dajejo kvantitativne ocene razlik v afinitetah med sorodnimi ligandi, kar je zelo uporabno pri vodeni preobrazbi spojin.
- Dodatno se uporabljajo metode za napoved ADMET lastnosti (absorpcija, porazdelitev, presnova, izločanje, toksičnost) in farmakoforni modeli za optimizacijo vodljivosti spojin.
Programska oprema in strojna oprema
Obstaja veliko paketov in orodij za različne naloge računalniške kemije. Nekatera znana imena vključujejo:
- Kvantno-mehanska orodja: Gaussian, ORCA, NWChem, Q-Chem.
- Molekulska dinamika: GROMACS, AMBER, CHARMM, NAMD.
- Dokiranje in virtualno preskušanje: AutoDock, Vina, Glide, GOLD.
- Orodja za analizo in vizualizacijo: VMD, PyMOL, Chimera.
Raste tudi uporaba grafičnih procesnih enot (GPU) za pospešitev MD in nekaterih kvantno-mehanskih izračunov ter uporaba računanja v oblaku in visokozmogljivostnih računalnikov (HPC). V prihodnosti bo kvantno računalništvo potencialno vplivalo na določene kvantno-mehanske izračune.
Validacija, omejitve in dobre prakse
Rezultate vedno velja preveriti z razpoložljivimi eksperimentalnimi podatki. Glavne omejitve so:
- Napake zaradi izbranih aproksimacij (npr. funkcional v DFT, parametri v silnih poljih).
- Omejitve časovnih in prostorskih merilnikov — nekatere procese je težko modelirati zaradi velikega obsega ali dolgih časovnih sklopov.
- Pomanjkljivosti v parametrih in prenosljivosti modelov med različnimi sistemih.
Dobre prakse vključujejo benchmarking metod za sorodne sisteme, preverjanje konvergence z različnimi osnovami ali parametri, ter kombiniranje računalniških napovedi z eksperimentalnimi meritvami.
Pogoste aplikacije
- Načrtovanje zdravil in ligantov, napoved afinitet ter optimizacija farmakokinetičnih lastnosti.
- Raziskave materialov: baterije, polprevodniki, katalizatorji, površinske lastnosti.
- Razumevanje mehanizmov kemijskih reakcij, katalize in procesa v biokemiji (encimi).
- Simulacije spektrov (IR, NMR, UV–Vis), kar pomaga pri interpretaciji eksperimentalnih podatkov.
Prihodnost: strojno učenje in nove tehnologije
V zadnjih letih se močno povečuje uporaba strojnega učenja (ML) za pospeševanje izračunov: od ML-potentialov, ki posnemajo kvantno-mehanske rezultate z veliko hitrostjo, do generativnih modelov za oblikovanje novih molekul. Hibridne metode, integracija opazovanj iz velikih podatkovnih nizov in razvoj kvantnega računalništva napovedujejo hitro napredujoče trajektorije v računalniški kemiji.
Kratki povzetek
Računalniška kemija je široko in hitro rastoče področje, ki povezuje teorijo, računalniške metode in eksperimentalne podatke za razumevanje in napovedovanje kemijskih pojavov. Z izbiro ustrezne metode, skrbno validacijo in kombinacijo več pristopov lahko raziskovalci učinkovito naslovijo vprašanja od temeljnih mehanizmov reakcij do praktičnega načrtovanja zdravil in naprednih materialov.

Sorodne strani
- Bioinformatika
- Statistična mehanika
Vprašanja in odgovori
V: Kaj je računalniška kemija?
O: Računalniška kemija je veja kemije, ki za reševanje kemijskih problemov uporablja računalništvo. Uporablja se lahko za izračun strukture in lastnosti molekul in trdnih snovi, napovedovanje kemijskih pojavov, ki še niso bili opaženi, ter načrtovanje novih zdravil in materialov.
V: Katere vrste sistemov obravnava računalniška kemija?
O: Računalniška kemija obravnava statične in dinamične sisteme. Sistem je lahko posamezna molekula, skupina molekul ali trdna snov.
V: Katere vrste informacij lahko zagotovi računalniška kemija?
O: Računalniška kemija lahko zagotovi informacije, kot so struktura (položaj atomov), absolutne in relativne energije, porazdelitev elektronskega naboja, dipoli in višji multipolni momenti, vibracijske frekvence, reaktivnost ali druge spektroskopske količine ter preseki za trke z drugimi delci.
V: Kako natančne so metode, ki se uporabljajo v računalniški kemiji?
O: Natančnost metod, ki se uporabljajo v računalniški kemiji, je od zelo natančnih do zelo približnih. Zelo natančne metode so običajno izvedljive le za majhne sisteme.
V: Kako računalniška kemija dopolnjuje eksperimentalne podatke?
O: Računalniška kemija običajno dopolnjuje informacije, pridobljene s kemijskimi poskusi. Uporablja se lahko za napovedovanje rezultatov, ki še niso bili eksperimentalno opaženi.
V: Ali velikost preučevanega sistema vpliva na to, koliko računalniškega časa potrebujemo?
O: Da - z velikostjo preučevanega sistema se povečuje tudi količina računalniškega časa, potrebnega za analizo, ter viri, kot sta pomnilnik in prostor na disku, potreben za shranjevanje.
Sorodni članki
Avtor
AlegsaOnline.com Računalniška kemija: metode, simulacije in načrtovanje zdravil Leandro Alegsa
URL: https://sl.alegsaonline.com/art/22297