Osteogenesis imperfecta: genetska bolezen krhkih kosti, simptomi in tipi
Osteogenesis imperfecta – genetska bolezen krhkih kosti: simptomi, tipi, pogosti zlomi, sprememba barve oči in izguba sluha. Celovit vodnik za razumevanje in podporo.
Osteogenesis imperfecta je genetska bolezen, znana tudi kot bolezen krhkih kosti. Najpogosteje je dedovana kot avtosomno dominantna motnja, kar pomeni, da zadostuje sprememba v enem od starševskih genov, da se bolezen pojavi, vendar so znani tudi primeri z avtosomno recesivno dednostjo ali s de novo (sprotnimi) mutacijami pri prizadetem posamezniku. Pri mnogih oblikah so prizadeti geni, ki kodirajo kolagen tipa I (najpogosteje mutacije v genih COL1A1 in COL1A2). OI prizadene del kosti, imenovan kolagenska palica, ki zagotavlja trdnost kosti. Nenormalni gen lahko oslabi strukturo kolagena ali zmanjša njegovo količino, kar povzroča povečano lomljivost kosti. Bolezen je prvi opisal Vrolik leta 1854.
Galerija slik
10 Slike







Klinični znaki in simptomi
Znaki se razlikujejo po resnosti, od blage povečane lomljivosti kosti do življenjsko nevarnih oblik. Pogosti simptomi vključujejo:
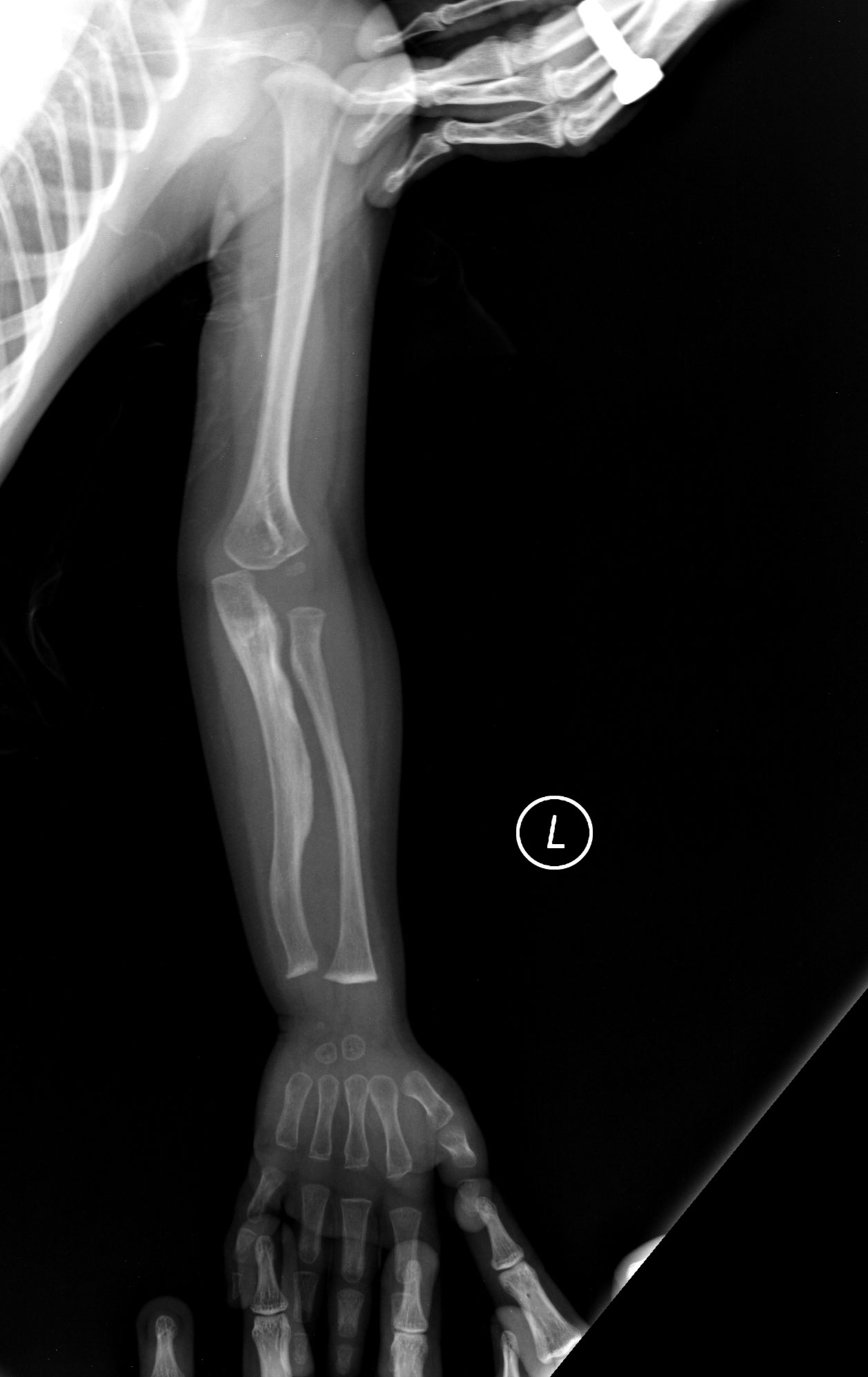
- Večkratni zlomi kosti ob minimalni ali nobeni travmi.
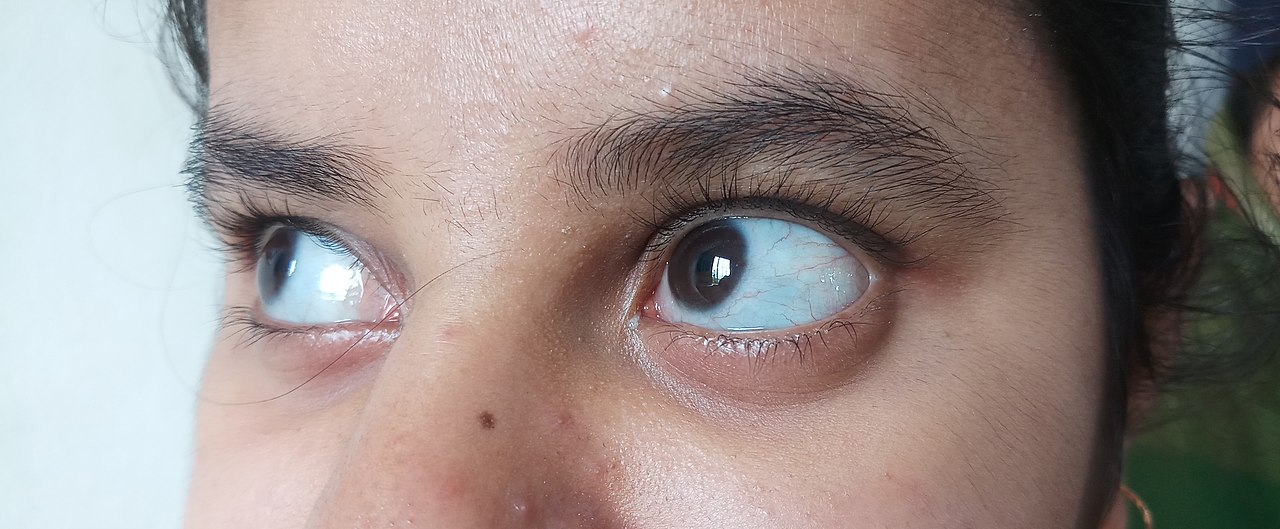
- Modrikaste (vijoličaste) očesne beločnice (sclera), kar je posledica tankosti vezivnega tkiva in vidne choroide.
- Dentinogeneza imperfecta – opalescentni, krhki zobje z večjim tveganjem za poškodbe in obrabo.
- Deformacije kosti (krivljenja nog, hrbtenice), krajša telesna višina pri hujših oblikah.
- Izguba sluha – pri približno 50 % odraslih z OI se razvije enostavna ali progresivna izguba sluha zaradi sprememb v srednjem ušesu ali notranjem ušesu.
- Skrajšana gibljivost, mišična šibkost in raztegljivost sklepov.
- V hujših oblikah lahko pride do dihalnih težav in zapletov pri novorojenčkih.
Tipi bolezni
Bolezen se klasično deli po Sillenceovi klasifikaciji na tipe I–IV, vendar je danes poznanih več kot 18 genetskih vrst OI. Najpogostejši so:
- Tip I – najblažja oblika: povečana nagnjenost k zlomom, modrikaste beločnice, pogosto normalna ali nekoliko zmanjšana rast, zobne spremembe lahko prisotne.
- Tip II – perinatalno oziroma novorojenjsko smrtno nevarna oblika: hude deformacije kosti, pogosto smrt v zgodnjem novorodstvenem obdobju zaradi okvar dihal in hudih zlomov.
- Tip III – huda, progresivno deformirajoča oblika: številni zlomi že pred puberteto, izrazite skeletne deformacije, pogosto izguba višine in težave s hrbtenico.
- Tip IV – zmerno huda oblika: simptomatologija med tipoma I in III, različna stopnja deformacij in lomljivosti.
Pomembno: nekateri zapisi v osnovnih virih poenostavijo razlage (npr. da je tip II le hujša različica tipa I). Dejstvo je, da ima vsak tip svojo klinično sliko in prognozo, pri čemer se spektar bolezni lahko med posamezniki močno razlikuje.
Diagnoza
- Klinična ocena: anamneza ponavljajočih se zlomov, družinska anamneza, značilni znaki (modre beločnice, zobne spremembe).
- Radiološke preiskave: rentgenski posnetki kosti, ki pokažejo tipične spremembe; DEXA (merjenje kostne gostote) za oceno osteopenije/osteoporoze.
- Genetsko testiranje: iskanje mutacij v genih, povezanih s kolagenom tipa I (npr. COL1A1, COL1A2) in drugih genih; pomembno za natančno diagnostiko in svetovanje.
- V nekaterih primerih se uporablja tudi biokemična analiza kolagena iz fibroblastov kože.
Zdravljenje in obvladovanje
OI ni ozdravljiva, a so na voljo ukrepi za zmanjšanje zlomov, obvladovanje bolečin in izboljšanje kakovosti življenja. Zdravljenje je multidisciplinarno in vključuje:
- Farmakoterapija: bisfosfonati (npr. pamidronat, zoledronska kislina) se uporabljajo pri otrocih in odraslih za večanje kostne gostote in zmanjšanje pogostosti zlomov.
- Fizioterapija in rehabilitacija: krepitev mišic, izboljšanje gibljivosti in učenje varnih načinov premikanja.
- Ortopedski posegi: intramedularni (čevasti) žeblji, korektivne osteotomije za stabilizacijo in zmanjšanje deformacij ter preprečevanje zlomov.
- Stomatološka oskrba: zdravljenje dentinogenih sprememb, zaščita zob, zgodnja intervencija pri otrocih.
- Slušni aparati in kirurški posegi: za izgubo sluha; v določenih primerih je možna tudi ušesna kirurgija ali implantacija kochlearnega vsadka.
- Preprečevanje poškodb, ustrezna prehrana, vključno z dovolj kalcija in vitamina D, ter redni pregledi specialistov.
Genetsko svetovanje in prognoza
Prognoza je zelo odvisna od tipa in resnosti bolezni: nekateri bolniki živijo dolgo in aktivno življenje z ustrezno obravnavo, medtem ko so najtežje oblike lahko usodne že v novorojenstvem obdobju. Genetsko svetovanje je ključno za družine z OI, saj omogoča razumevanje načina dedovanja, tveganj za potomce in možnosti prenatalnega diagnosticiranja ali preimplantacijske genetske diagnostike (PGD), kadar je znana patogena mutacija.
Sklep: Osteogenesis imperfecta je spekter genetskih motenj kolagena, ki povzročijo povečano lomljivost kosti in spremljajoče sistemske težave. Z zgodnjo diagnozo, individualiziranim zdravljenjem in celostno oskrbo lahko mnogi bolniki dosežejo boljšo kakovost življenja in zmanjšajo zaplete.
Simptomi
Manj hudi simptomi OI lahko vključujejo:
- zlahka zlomljene kosti
- zrahljani sklepi
- nizek mišični tonus.
- modra, vijolična ali siva barva običajno belega dela oči.
- trikotna oblika obraza
- nagnjenost k razvoju skolioze.
- krhki zobje
OI ima številne druge resne in smrtonosne simptome, vključno s težavami z dihanjem in deformacijo kosti.
Demografski podatki
Vprašanja in odgovori
V: Kaj je osteogenesis imperfecta?
O: Osteogenesis imperfecta je genetska motnja, ki jo običajno imenujemo bolezen krhkih kosti. Oslabi ali uniči kolagensko palico, ki zagotavlja trdnost kosti, in povzroči, da se kosti pogosteje lomijo.
V: Kako se deduje osteogenesis imperfecta?
O: Osteogenesis imperfecta je običajno avtosomno dominantna bolezen, kar pomeni, da jo lahko oseba dobi, če ima samo eden od staršev nenormalen gen.
V: Kdo je prvi ugotovil osteogenesis imperfecta?
O: Vrolik je leta 1849 prvi ugotovil osteogenesis imperfecta.
V: Ali obstaja zdravilo za osteogenesis imperfecta?
O: Na žalost osteogenesis imperfecta ni ozdravljiva.
V: Katere so štiri vrste osteogenesis imperfecta?
O: Štiri vrste osteogenesis imperfecta so tip 1, tip 2, tip 3 in tip 4.
V: Kateri so simptomi osteogeneze imperfekcije tipa tri?
O: Osebe s tretjim tipom osteogenesis imperfecta imajo lahko pred puberteto več kot 100 zlomov. Njihove oči pogosto dobijo vijoličast, moder ali siv odtenek, ljudje s tem primerom pa imajo pogosto tudi izgubo sluha.
V: Kako pogosta je izguba sluha pri osebah z osteogenesis imperfecta?
O: 50 % ljudi z osteogenesis imperfecta ima v odrasli dobi izgubo sluha.
Sorodni članki
Avtor
AlegsaOnline.com Osteogenesis imperfecta: genetska bolezen krhkih kosti, simptomi in tipi Leandro Alegsa
URL: https://sl.alegsaonline.com/art/73422
Viri
- oif.org : "Osteogenesis Imperfecta Foundation:"
- nlm.nih.gov : "Autosomal dominant: MedlinePlus Medical Encyclopedia"
- oif.org : "Osteogenesis Imperfecta Foundation: Understanding Bone Structure"