Cistična fibroza (mukoviscidoza): definicija, genetika, simptomi, zdravljenje
Cistična fibroza (mukoviscidoza): jasna definicija, genetski vzroki, ključni simptomi in sodobne možnosti zdravljenja ter življenjski nasveti za bolnike in družine.
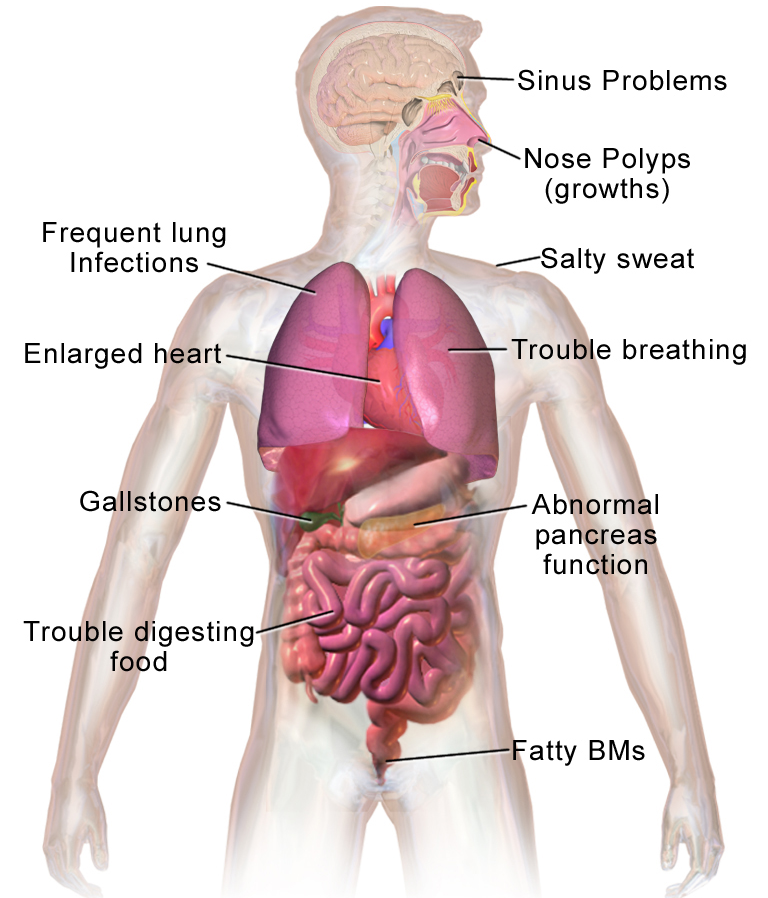
Cistična fibroza, znana tudi kot mukoviscidoza, CF, je dedna bolezen, pri kateri telo proizvaja gosto, lepljivo sluz, ki se nabira v pljučih, prebavnem sistemu in drugih delih telesa. Nabiranje sluzi ovira dihanje, spodbuja ponavljajoče se okužbe in lahko ovira presnovo hrane v črevesju. Gre za sistemsko bolezen, ki najbolj prizadene dihala in prebavila, vendar lahko vpliva tudi na jetra, reprodukcijo in rast.
Galerija slik
4 Slike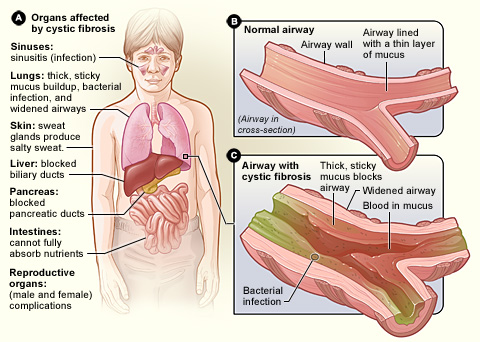

Genetika in prenos
Cistično fibrozo povzroča mutacija v genu, imenovanem CFTR. Če imata oba starša mutiran gen za cistično fibrozo (prenašalca), obstaja možnost, da otrok podeduje bolezen. Bolezen se prenaša avtosomno recesivno, kar pomeni:
- Če sta oba starša prenašalca: 25 % verjetnosti, da bo otrok bolan, 50 % da bo nosilec brez simptomov in 25 % da ne bo nosilec.
- Če je le en starš prenašalec: otrok ne bo bolan, lahko pa bo prenašalec.
Starš morda nima simptomov, vendar lahko nosi mutacijo. Oseba s cistično fibrozo ni nalezljiva (ne prenaša bolezni na druge z okužbo). Obstaja veliko različnih mutacij v CFTR genu; nekatere so pogostejše (npr. ΔF508), druge redkejše, zato se klinični potek in resnost bolezni razlikujeta.
Znaki in simptomi
Simptomi se lahko pojavijo že novorojenčku ali pozneje v otroštvu oziroma odrasli dobi. Najpogostejši znaki vključujejo:
- Dihala: kronični produktivni kašelj, ponavljajoče se pljučne okužbe, piskanje, zmanjšana telesna vzdržljivost in v hujših primerih bronhiektazije ali krvavitev iz dihal.
- Prebava: težave s prebavo zaradi pomanjkanja encimov trebušne slinavke (pankreasna insuficienca) – maščobna diareja, počasna rast, slabša teža in pri novorojenčkih lahko mekonijev ileus (zamašitev črevesja).
- Reproduktivni sistem: pri moških pogosto neplodnost zaradi neprehodnih semenovodov (azoospermija, CBAVD), ženske imajo lahko zmanjšano plodnost zaradi goste cervikalne sluzi.
- Druge težave: diabetes, jetrna bolezen z obstrukcijo žolčnih kanalov, osteoporoza, visok koncentrat soli v znoju kar lahko vodi do dehidracije in neravnovesja elektrolitov.
Diagnoza
Diagnoza cistične fibroze temelji na kombinaciji kliničnih znakov in specifičnih testov. Pogosti diagnostični postopki so:
- Novorojensko presejanje (screening) – v mnogih državah rutinski test pri novorojenčkih.
- Test znoja (klasičen test) – merjenje klora v znoju; visoke vrednosti podpirajo diagnozo.
- Genetsko testiranje – iskanje mutacij v CFTR genu za potrditev diagnoze in določitev tipa mutacije.
- Fiziološki testi dihal (npr. pljučne funkcije) in morebitne slikovne preiskave (RTG, CT) za oceno stanja pljuč.
Zdravljenje in zdravljenje simptomov
Za cistično fibrozo ni univerzalnega zdravila, vendar so na voljo številne terapije, ki bistveno izboljšajo kakovost življenja in pričakovano življenjsko dobo. Cilj zdravljenja je zmanjševanje simptomov, preprečevanje zapletov in ohranjanje pljučne funkcije.
- Fizioterapija dihal: redno čiščenje dihal z manualnimi tehnikami ali napravami za odstranjevanje sluzi.
- Difikatorji sluzi in inhalacije: dornaza alfa, inhalacije slane vode (hypertonic saline) in bronhodilatatorji pomagajo redčiti in odstraniti sluz.
- Antibiotiki: za zdravljenje in preprečevanje pljučnih okužb; v nekaterih primerih inhalirani antibiotiki (npr. tobramicin) za kronične okužbe s Pseudomonas aeruginosa.
- V nadomestno encimsko terapijo: nadomestna terapija pankreasnih encimov pri tistih z insuficienco trebušne slinavke, skupaj z dieto z višjim vnosom kalorij in dopolnjevanjem vitaminov topnih v maščobah (A, D, E, K).
- CFTR modulatorji: ciljne zdravila, ki popravljajo delovanje okvarjenega proteina CFTR pri določenih mutacijah (npr. ivacaftor, kombinacije kot so lumacaftor/ivacaftor, tezacaftor/ivacaftor in elexacaftor/tezacaftor/ivacaftor). Ta zdravila so revolucionirala zdravljenje za mnoge bolnike z ustreznimi genetskimi mutacijami.
- Ljudje z napredovalo boleznijo: pri hudih zapletih pljučne funkcije je lahko opcija presaditev pljuč.
- Podpora in preventivni ukrepi: redni zdravniški pregledi v centru za CF, cepljenja, izogibanje izpostavljenosti okužbam in ukrepi za preprečevanje medsebojne kontaminacije med bolniki s CF.
Življenje z cistično fibrozo
S sodobnim celostnim zdravljenjem mnogi ljudje s cistično fibrozo živijo dlje in bolj polno življenje. Pomembna je multidisciplinarna obravnava – pulmonolog, gastroenterolog, dietetik, fizioterapevt, genetik in psiholog. Redna telesna aktivnost, ustrezna prehrana, skrb za zobje in mentalno zdravje ter upoštevanje zdravniških navodil znatno pripomorejo k boljšemu izidu.
Genska svetovanja in preprečevanje
Če obstaja družinska anamneza CF ali če par razmišlja o otrocih, je priporočljivo genetsko svetovanje in testiranje prenašalcev. Prenatalno testiranje in predimplantacijska genetska diagnostika sta možnosti za pare, ki želijo zmanjšati tveganje za prenos bolezni.
Čeprav zdravila za cistično fibrozo ni v splošnem pomenu popolnega zdravila, obstaja veliko zdravil, ki pomagajo ohranjati zdravje, zmanjševati simptome in podaljševati življenje. Ob sumu na cistično fibrozo ali pri sladnjem testu novorojenčka se obrnite na specialista za natančno diagnostiko in načrt obravnave.
Kako CF vpliva na telo
Cistična fibroza prizadene celotno telo. Na splošno ima telo težave s prenosom soli v dele telesa, ki jo potrebujejo. Ker ima telo težave s premikanjem soli, se ta kopiči na mestih, kamor ne bi smela, na primer v pljučih, želodcu in črevesju.
Pljuča
Ko se v pljučih zatakne sol, je v njih manj vode, zato postane sluz zelo gosta. Zelo težko je dihati. To zdravimo z zdravili za dihanje, ki pomagajo dodajati vodo v pljuča, da je sluz redkejša in jo je lažje izkašljevati. Ko je sluz redkejša in manjša, je lažje dihati.
Zdravljenje
Zdravila za cistično fibrozo ni. Čeprav lahko ljudje naredijo marsikaj, da ostanejo zdravi. Zdrave navade preprečujejo, da bi oseba še bolj zbolela. Ljudje lahko ostanejo čisti. Ljudje se lahko izogibajo mikrobom. Lahko pijejo vodo, ki pomaga pri izločanju sluzi. Jemanje encimov pomaga pri prebavi hrane, če je v želodcu sluz.
Vadba čisti sluz. Gradi močne mišice in kosti ter krepi pljuča. Jemanje vitaminov pomaga telesu v boju proti okužbi. Prav tako pomaga telesu pri rasti in dobrem delovanju.
- Inhalacijski antibiotiki se uporabljajo za preprečevanje rasti bakterij v gosti sluzi.
- Vdihavanje slane vode pomaga pri vlaženju pljuč.
Testiranje za cistično fibrozo
- Kloridni test znoja - z njim se preveri vsebnost soli v znoju.
- Genetski test - uporabi se, če je test z znojem pozitiven, da se ugotovi, ali imajo oba gena.
65 vrtnic
Nekateri otroci svoje stanje označujejo z besedo "65 vrtnic", saj je cistična fibroza za majhnega otroka težko izgovorljiva. "65 vrtnic" je tudi besedna zveza z zaščiteno blagovno znamko Fundacije za cistično fibrozo, ki pomaga nadzorovati njeno uporabo. To je zelo koristen način za razumevanje pri majhnih otrocih. Ko ga izgovorimo na glas, zveni podobno kot cistična fibroza.
Vprašanja in odgovori
V: Kaj je cistična fibroza?
O: Cistična fibroza je bolezen, zaradi katere telo proizvaja gosto, lepljivo sluz, ki se lahko nabira v pljučih, prebavnem sistemu in drugih delih telesa.
V: Kako nastane cistična fibroza?
O: Cistično fibrozo povzročita oba starša, ki sta podedovala gen za cistično fibrozo.
V: Ali lahko oseba z enim samim genom za cistično fibrozo bolezen prenese na otroka?
O: Oseba z enim samim genom za cistično fibrozo morda sama nima bolezni, vendar jo lahko prenese na svojega otroka.
V: Ali je cistična fibroza nalezljiva?
O: Ne, cistična fibroza ni nalezljiva in se ne more prenašati z ene osebe na drugo.
V: Ali obstaja zdravilo za cistično fibrozo?
O: Ne, za cistično fibrozo trenutno ni zdravila, obstajajo pa številna zdravila, ki lahko pomagajo pri obvladovanju bolezni.
V: Katere dele telesa lahko prizadene cistična fibroza?
O: Cistična fibroza lahko prizadene pljuča, prebavni sistem in druge dele telesa.
V: Kako lahko ljudje s cistično fibrozo ostanejo zdravi?
O: Ljudje s cistično fibrozo lahko ostanejo zdravi, če jemljejo zdravila, spremljajo svoje stanje in živijo zdravo.
Sorodni članki
Avtor
AlegsaOnline.com Cistična fibroza (mukoviscidoza): definicija, genetika, simptomi, zdravljenje Leandro Alegsa
URL: https://sl.alegsaonline.com/art/24952