Creutzfeldt-Jakobova bolezen (CJD): kaj je, vzroki in simptomi
Creutzfeldt-Jakobova bolezen (CJD): odkrijte vzroke prionov, zgodnje simptome, potek in ključne informacije za razumevanje in prepoznavanje te smrtonosne nevrodegenerativne bolezni.
Creutzfeldt-Jakobova bolezen (izgovori se KROITS-felt YAH-kohb) ali CJD je nevrološka bolezen. Je degenerativna (sčasoma se poslabša), ni je mogoče ozdraviti in vedno povzroči smrt. CJD se včasih imenuje človeška oblika "bolezni norih krav" (goveja spongiformna encefalopatija ali BSE). BSE je pravzaprav vzrok ene redke vrste Creutzfeldt-Jakobove bolezni; ne gre za isto bolezen.
CJD povzroča infekcijskiagens, imenovan prion. Prioni so beljakovine, ki so napačno zložene. Prioni naredijo svoje kopije tako, da pravilno zložene beljakovine spremenijo v napačno zložene oblike. CJD povzroči, da možgansko tkivo zelo hitro postane nezdravo. Ko bolezen uničuje možgane, se v možganih pojavijo luknje. Struktura možganov se spremeni in postane podobna kuhinjski gobi.
Galerija slik
6 Slike




Vrste CJD
Obstaja več oblik CJD, ki se razlikujejo po vzroku:
- Sporadična CJD (sCJD) – najpogostejša oblika; pojavi se nenadoma brez znanega vzroka.
- Dedna (familialna) CJD (fCJD) – povezana z mutacijami v genu PRNP, prenos med družinskimi člani po dedovanju.
- Variante CJD (vCJD) – redka oblika, povezana s uživanjem mesa okuženega z BSE; običajno prizadene mlajše osebe in poteka drugače kot sCJD.
- Iatrogena CJD – redka prenos zaradi medicinskih postopkov (npr. okuženi instrumenti, presaditve tkiv, prejeti humani rastni hormon).
Vzroki in prenos
CJD povzroča prion — nenormalna različica normalne beljakovine (PrP). Prioni so zelo odporni na običajne postopke razkuževanja in lahko trajajo v okolju ali na medicinski opremi. Prenos je redek; večina primerov je sporadičnih ali dednih. Zabeleženi načini prenosa vključujejo:
- dedne mutacije v genu PRNP,
- izpostavljenost okuženim tkivom (npr. pri kirurških posegih ali presaditvah),
- uživanje izdelkov iz mesa, okuženih z BSE (povezano z vCJD).
Znaki in simptomi
Simptomi običajno napredujejo zelo hitro (v urah do mesecih). Med najpogostejšimi znaki so:
- hitro napredujoča demenca (izguba spomina, zmanjšana miselna zmogljivost),
- spremembe osebnosti (anksioznost, depresija, razdražljivost),
- motnje gibanja in koordinacije (ataksija), težave z hojo, nezmožnost izvedbe zaporednih gibov,
- mioklonus — nenadzorovani »skočni« gibi mišic, pogosto sproženi z zvokom ali dotikom,
- govorne in vidne motnje (zamaščen govor, dvojni vid),
- napredna faza: težave z hranjenjem, okužbami, komo.
Diagnoza
Diagnoza temelji na kliničnih znakih, slikovnih preiskavah in posebnih testih. Ker je bolezen redka in resna, se diagnoza običajno postavi v specializiranih centrih. Pogoste preiskave vključujejo:
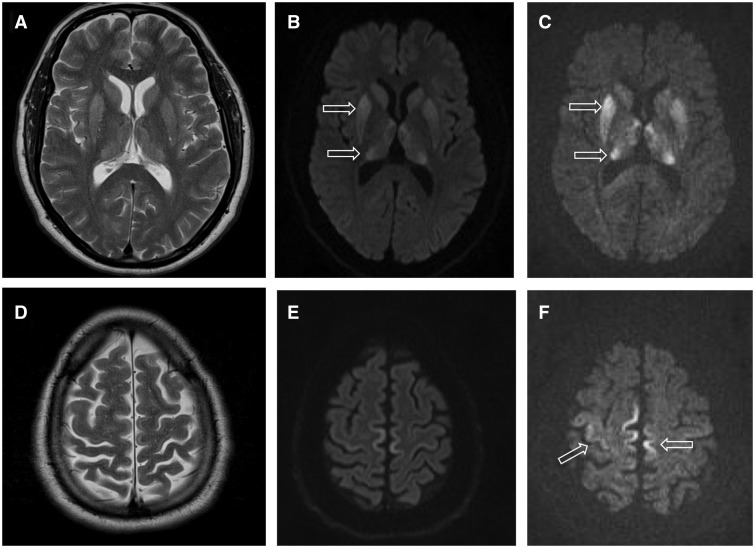
- magnetna resonance (MRI) možganov — značilne spremembe v nekaterih predelih možganov,
- elektroencefalogram (EEG) — lahko pokaže specifične vzorce pri sCJD,
- analiza cerebrospinalne tekočine (CSF) — testi, kot so 14-3-3 protein in sodobnejši RT-QuIC, pomagajo podpreti diagnozo,
- v nekaterih primerih biopsija ali obdukcija — za potrditev s histologijo in detekcijo prionov.
Zdravljenje in oskrba
Za CJD ni zdravila, ki bi bolezen pozdravilo ali prekinilo njeno napredovanje. Zdravljenje je podporno in usmerjeno v izboljšanje kakovosti življenja ter lajšanje simptomov:
- zdravila proti bolečinam, za nadzor mioklonusa ali za obvladovanje anksioznosti in depresije,
- fizioterapija in delovna terapija za ohranitev gibljivosti in funkcij čim dlje,
- podpora hranjenju (npr. s sondeo) in paliativna oskrba v napredovalih fazah,
- skrb za varnost pacienta zaradi padcev in nenadzorovanih gibov.
Prognosis
CJD običajno napreduje hitro. Večina ljudi umre v nekaj mesecih do dveh let od začetka simptomov, pri čemer je mediana preživetja pri sCJD približno 4–6 mesecev. vCJD lahko traja nekoliko dlje, vendar je tudi običajno usodna.
Preprečevanje in javnozdravstveni ukrepi
Ker so prioni odporni in prenos možen pri nekaterih medicinskih postopkih, obstajajo strogi standardi za sterilizacijo in ravnanje z visokorizičnim materialom. Javne zdravstvene ukrepe vključujejo nadzor nad dobavo mesa (za preprečevanje BSE) in prepovedi uporabe visokorizičnih tkiv pri presaditvah ali donorstvu. Krvni donorji so pogosto predmet omejitev, kadar obstaja tveganje izpostavljenosti vCJD.
Kdaj poiskati zdravniško pomoč
Če se pri vas ali pri bližnjem pojavi hitro napredujoča slabost spomina, nenavadne mišične trzljaje, hitro poslabšanje mišljenja ali hude vedenjske spremembe, je pomembno, da čim prej obiščete zdravnika. Zgodnja ocena omogoči ustrezno diagnostiko, podporo in načrtiranje oskrbe.
Čeprav je CJD redka, so njeni učinki hudi. Zdravniki, nevrologi in javnozdravstvene službe sodelujejo, da bi preprečili prenos in nudili podporo bolnikom ter njihovim družinam.
Vrste in vzroki CJD
Vrste CJD vključujejo:
- (vCJD):
To vrsto CJD lahko povzroči uživanje hrane, v kateri so prioni, na primer mesa krav, ki so zbolele za BSE ("bolezen norih krav"). Vendar je to zelo redek vzrok CJD.
- sporadična (sCJD):
To je najpogostejša vrsta CJD. V 85 % primerov CJD gre za sporadično CJD. Nihče ne ve, kaj povzroča sCJD; zdi se, da se pojavlja naključno.
- družinska (fCJD):
Večina od preostalih 15 % primerov CJD je družinska CJD. To je oblika CJD, ki se pojavlja v družinah.
- iatrogeni:
To obliko CJD običajno povzroči medicinski postopek, pri katerem oseba dobi kri ali tkivo osebe s CJD. Oseba lahko na primer dobi iatrogeno CJD, če dobi transfuzijo krvi ali presaditev roženice od osebe, ki ima CJD.
Znaki in simptomi
Prvi simptom CJD je demenca, ki se zelo hitro poslabša. demenca povzroča izgubo spomina, osebnostne spremembe in halucinacije.
Drugi pogosti duševni simptomi so:
- Anksioznost
- Depresija
- Paranoja
- Obsesivno-kompulzivni simptomi
- Psihoza
Fizični simptomi CJD pogosto vključujejo:
- Težave z govorjenjem
- Trmasti gibi (mioklonus)
- Težave z ravnotežjem (ataksija)
- Težave pri hoji
- Tresenje ali togost
- Težave z vidom
- težave s požiranjem, ki lahko otežijo ali onemogočijo prehranjevanje.
- težave s kašljem, ki lahko povzročijo pljučnico
- gibi, ki jih bolnik ne more nadzorovati (diskinezija).
Večina ljudi s CJD umre v šestih mesecih po pojavu prvih simptomov. Pogosto umrejo zaradi pljučnice, ki jo povzročijo težave s kašljem. Približno 15 % bolnikov preživi dve leti ali več. Nekateri bolniki živijo 4-5 let s pretežno duševnimi simptomi, dokler se bolezen ne poslabša in povzroči več telesnih simptomov. Ko se to zgodi, ljudje običajno umrejo v enem letu.
Simptomi CJD so posledica odmiranja vedno večjega števila možganskih živčnih celic. Ko znanstveniki pod mikroskopom pregledajo možgansko tkivo bolnika s CJD, lahko vidijo številne drobne luknjice, v katerih so odmrla cela področja živčnih celic.
Diagnoza
Zdravniki lahko posumijo na CJD, kadar ima oseba določene simptome. Demenca se na primer običajno slabša počasi. Demenca, ki se zelo hitro poslabša, je nenavadna. Skupaj s simptomi, kot so surovi gibi, lahko ti simptomi kažejo na možnost CJD.
S testi se lahko ugotovi, ali ima oseba CJD. Ti testi vključujejo:
- Elektroencefalografija (EEG): Ta preiskava pokaže električno aktivnost v možganih. Zdravnik lahko pogosto opazi spremembe v EEG, ki so značilne za osebe s CJD. Vrsta sprememb, ki se pokažejo na EEG, je odvisna od vrste CJD, ki jo ima bolnik, in od tega, kako daleč je njegova bolezen.
- Lumbalna punkcija (spinalna punkcija): S tem testom je mogoče preučiti cerebrospinalno tekočino (tekočino, ki obdaja možgane in hrbtenjačo) ter poiskati specifično beljakovino ("beljakovina 14-3-3").
- MRI možganov: Preiskava, pri kateri z zelo močnim magnetom posnamemo slike možganov.
- Biopsija: Pri biopsiji kirurg z iglo odvzame majhen košček tkiva iz telesa, da ga lahko zdravniki pregledajo pod mikroskopom. vCJD se lahko diagnosticira z biopsijo mandeljnov. Za vse druge vrste CJD je biopsija možganov edini način, da se z gotovostjo ugotovi, ali ima oseba CJD. Ker pa biopsija možganov lahko povzroči poškodbe možganov, se običajno ne opravi, če so druge preiskave že pokazale, da ima oseba verjetno CJD.

Zdravljenje
Do leta 2016 še ni zdravila, ki bi ozdravilo CJD ali celo upočasnilo njene učinke. Opravljajo se številni poskusi, da bi našli načine zdravljenja.
Danes so edini način zdravljenja CJD zdravila, ki zdravijo simptome bolezni in pomagajo bolnikom, da se bolje počutijo. Bolniki, ki imajo napade, lahko na primer dobijo antikonvulzivna zdravila. Benzodiazepini lahko zmanjšajo pogostost mišičnih trzljajev.
Bolniki se lahko odločijo tudi za medicinske postopke, ki pomagajo pri odpravljanju slabih simptomov. CJD lahko na primer povzroči takšne težave pri požiranju, da oseba ne more jesti. Nekatere osebe s CJD se odločijo za vstavitev cevke za hranjenje, ko ne morejo več jesti. To je cevka, ki gre v želodec, tako da se lahko posebna tekočina daje neposredno v želodec, da se oseba nahrani.
Sorodne strani
- Prion
- Prionska bolezen
- Terminalna bolezen
Vprašanja in odgovori
V: Kaj je Creutzfeldt-Jakobova bolezen?
O: Creutzfeldt-Jakobova bolezen (CJD) je nevrološka bolezen, ki je degenerativna, neozdravljiva in vedno smrtna.
V: Ali obstaja zdravilo za CJD?
O: Ne, zdravila za CJD ni.
V: Zakaj se CJD včasih imenuje človeška oblika bolezni norih krav?
O: CJD se včasih imenuje človeška oblika "bolezni norih krav", ker je goveja spongiformna encefalopatija (BSE), ki je vzrok ene redke vrste CJD, splošno znana kot "bolezen norih krav".
V: Kaj je vzrok CJD?
O: CJD povzroča povzročitelj, imenovan prion, ki je nepravilno zložena beljakovina, ki lahko ustvarja svoje kopije, tako da spreminja pravilno zložene beljakovine v nepravilno zložene.
V: Kaj se zgodi z možganskim tkivom pri CJD?
O: CJD povzroči, da možgansko tkivo zelo hitro postane nezdravo, kar povzroči nastanek lukenj v možganih in spremembo strukture možganov, ki postanejo podobni kuhinjski gobi.
V: Ali je BSE ista bolezen kot CJD?
O: Ne, BSE ni ista bolezen kot CJD; dejansko je vzrok ene redke vrste CJD.
V: Kako prioni povzročajo CJD?
O: Prioni povzročajo CJD, ker se nepravilno zlagajo in ustvarjajo svoje kopije na račun pravilno zloženih beljakovin v možganih. To povzroči uničenje zdravega možganskega tkiva in nastanek lukenj, značilnih za bolezen.
Sorodni članki
Avtor
AlegsaOnline.com Creutzfeldt-Jakobova bolezen (CJD): kaj je, vzroki in simptomi Leandro Alegsa
URL: https://sl.alegsaonline.com/art/24152
Viri
- cdc.gov : "CJD (Creutzfeldt–Jakob Disease, Classic)"
- ncbi.nlm.nih.gov : "Creutzfeldt–Jakob disease: Transmissible spongiform encephalopathy; vCJD; CJD; Jacob-Creutzfeldt disease"
- bmj.com : "Bovine spongiform encephalopathy and variant Creutzfeldt–Jakob disease"
- doi.org : 10.1101/SQB.1996.061.01.052
- pubmed.ncbi.nlm.nih.gov : 9246478
- www3.interscience.wiley.com : "MM2-cortical-type sporadic Creutzfeldt–Jakob disease with early stage cerebral cortical pathology presenting with a rapidly progressive clinical course"
- doi.org : 10.1111/j.1440-1789.2008.00904.x
- pubmed.ncbi.nlm.nih.gov : 18410280
- who.int : who.int: "Fact sheets no 180: Variant Creutzfeldt-Jakob disease" Feb 2012 ed.
- prion.ucl.ac.uk : "Sporadic Prion Disease"
- doi.org : 10.1056/NEJM198610163151605
- pubmed.ncbi.nlm.nih.gov : 3762620
- prion.ucl.ac.uk : "Sporadic Prion Disease"
- merckmanuals.com : "Creutzfeldt–Jakob Disease (CJD)"
- ncbi.nlm.nih.gov : "Creutzfeldt-Jakob disease: Updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy"